Engineering selective infection
How viruses infect cells is key to developing safe oncolytic viral therapies. In this post we discuss how viruses can be engineered to target cancer cells.
The infection process begins when a virus recognizes and attaches to a host cell. This attachment is typically mediated by viral proteins known as glycoproteins, which specifically bind to receptors on the surface of the target cell. These glycoproteins act like keys, unlocking the door to the cell by interacting with specific cell surface receptors.
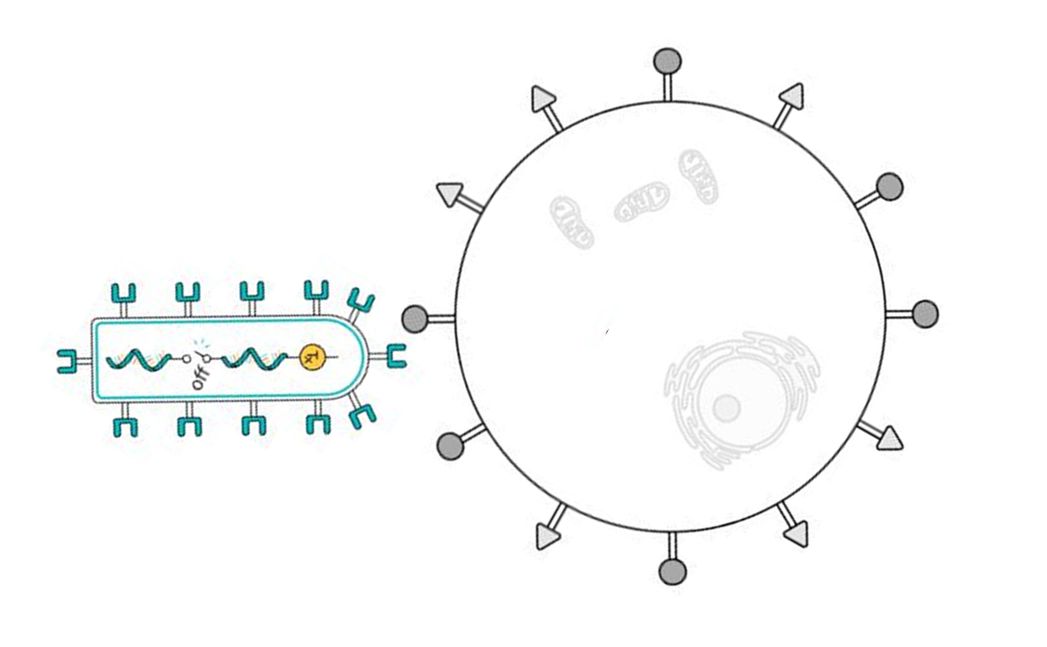
Once the virus has attached to the cell surface, the next step is entry. This can occur through direct fusion of the viral envelope with the cell membrane, facilitated again by the receptor glycoproteins. Alternatively, the virus can be taken into the cell via endocytosis, a process where the cell engulfs the virus in a bubble-like vesicle. Inside the cell, the virus releases its genetic material into the host cell’s cytoplasm.
This genetic material then hijacks the cell’s machinery to begin producing viral components. The host cell, now a viral factory, assembles these components into new virus particles. We have engineered viruses to do this only conditionally using aptazymes. (Read more on how to regulate viral replication using aptazymes )
In this post we focus on how our understanding of viral infection can be used to engineer selectively infective viruses to make safe oncolytic viral therapies.
How to retarget viruses
Different viruses target different receptors. SARS-CoV2 the ACE2 receptor, Seneca Valley Virus the TEM-8 receptor and Vesicular Stomatitis Virus the LDL receptor to name a few.
Retargeting viruses to infect cancer cells is an innovative approach in cancer therapy. Since most natural viruses do not inherently target cancer cells, genetic engineering is employed to create chimeric viruses that can selectively infect and destroy cancer cells.
What are some of the methods to retarget viruses, to make them more cancer specific?
Glycoprotein Substitution
One method is to replace the natural glycoprotein of a virus with that of another virus. This involves swapping the genetic sequence coding for the virus’s glycoprotein with that from a different virus that has an affinity for receptors overexpressed on cancer cells. For example, a virus that typically targets the LDL receptor might be modified to express a glycoprotein from Seneca Valley Virus (SVV) that binds to TEM-8, which is overexpressed in certain types of cancer cells.

The problem of this approach is the lack of flexibility, since we need to rely on naturally occurring viruses.
Insertion of Cancer-Specific Ligands
Another strategy involves genetically engineering the viral surface proteins to include ligands that specifically bind to receptors or molecules overexpressed on cancer cells. First, the glycoprotein is “de-targeted”. This means that the sequence is mutated so that natural infectivity is removed. Then, the glycoprotein is “retargeted”, whereby a cancer-specific ligand is inserted in the glycoprotein sequence. Here, the ligand protein is essentially fused on the outside of the viral glycoprotein. The goal is to re-enable infection, but for a target receptor of interest. These ligands can be partial proteins, antibodies, nanobodies, to name a few. For example, inserting antibodies that have a high affinity for receptors like HER2 or HER3. This approach is much more promising as with the right mutated glycoprotein hundreds of nanobodies can be made to work. Some groups use the measles virus glycoprotein to do this. We have done this successfully too.

In this figure we show an example from our more current work, where we have replaced the glycoprotein of VSV with the glycoprotein of Sindbis virus and added a GPC3 targeting nanobody.
Key requirements for selective infection
Firstly, it’s crucial to minimize off-target infectivity. We need to ensure that the engineered glycoprotein does not retain the ability to target its natural receptor, as this receptor may be present on healthy cells, potentially leading to off-target toxicity.
Another aspect is to minimize seroprevalence. The ideal scenario involves using a glycoprotein with no existing seroprevalence. While there is extensive literature on attaching ligands or nanobodies to the measles glycoprotein, most people have robust immune responses to measles, which could pose a challenge.
The size of the genetic code also plays a vital role. Highly lytic RNA viruses usually have short RNA sequences, limiting the space available to add the genetic code of the new glycoprotein, especially if it is significantly larger.
Finally, stability in the bloodstream is paramount. The new glycoprotein should demonstrate excellent stability, particularly considering the high temperatures encountered in the bloodstream.
Our accomplishments
Our initial work focused on answering the question whether we could retarget our base virus Vesicular Stomatitis Virus (VSV) to more specific targets. After that we started to use the second approach of using nanobodies.
We have found that it is possible to retarget VSV using a glycoprotein from a different virus. We have used the glycoproteins of Seneca Valley Virus, HCV, Measles, Sindbis and others successfully. Not only do the new chimeric viruses replicate, they replicate roughly at the same rate as the wild type VSV.
Our first work with nanobodies was based on the Measles glycoprotein, but we realized that it would not be as effective since there is widespread seroprevalence for Measles and also the sequence is quite long. We switched to using Sindbis for several reasons. It has no seroprevalence, its genomic sequence is short and it is stable at higher temperatures. We have made several functional viruses targeting HER2, GPC3, EGFR and other cancer relevant receptors.
Here we show the results of an infection test. We compare the selectivity of wild-type VSV with an engineered version of VSV targeting GPC3. The red dots are fluorescent proteins indicating infection of cells. It is clear that VSV wild-type infects both the healthy and the HepG2 cancer cells. The retargeted VSV only infects the HepG2 cells. 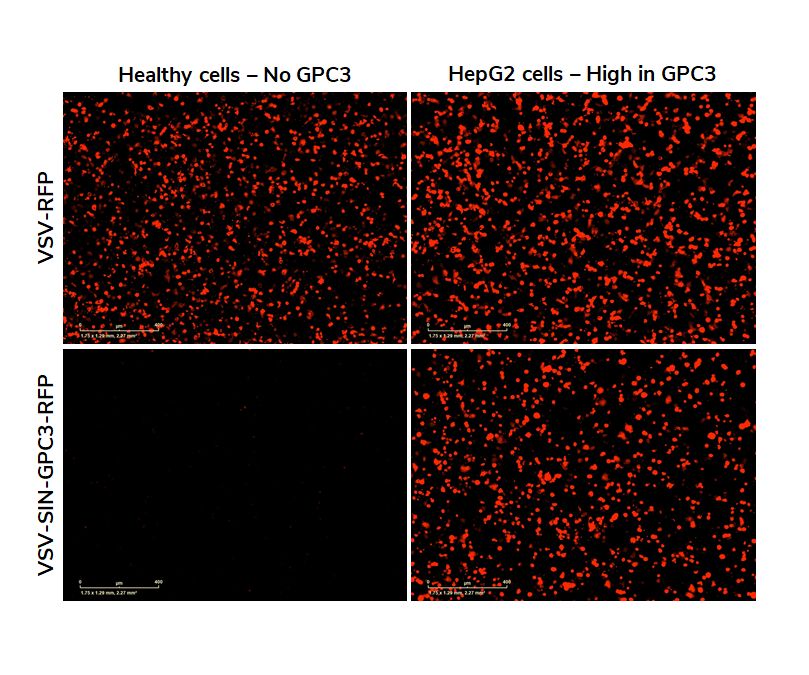
Most recently we are using different glycoproteins because of their superior lack of off target infections and stability characteristics.
One thought on “Engineering selective infection”
That is quite an accomplishment! Awesome work!